|
|
Sebastian-Platelet-Syndrom
Sebastian-Syndrom
Autor/en: F.-G. Hagmann
Institution/en: Karlsruhe
Serie zuletzt geändert am: 28.11.2011
|
Danksagung: Gewidmet Ralph M. Loreth
|
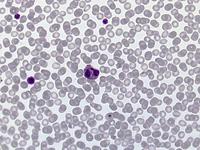
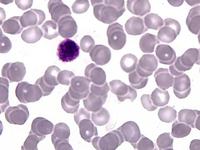
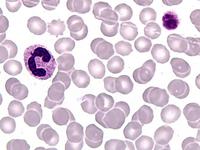
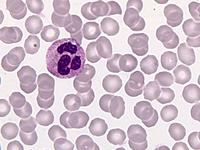
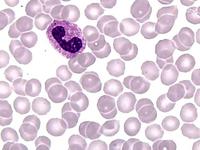
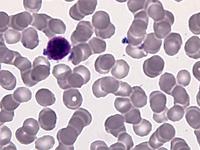
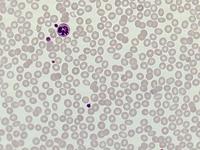
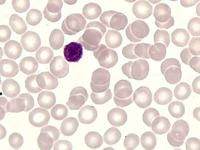
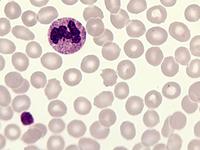
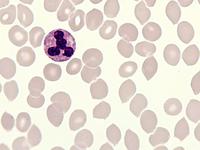
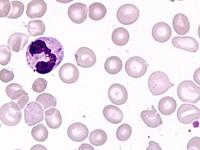
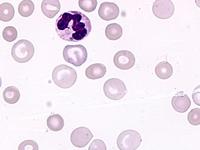
Das Sebastian-Platelet-Syndrom ist eine sehr seltene angeborene Thrombozytenstörung mit Makrothrombozyten, Thrombozytopenie und Einschlüssen im Zytoplasma der Neutrophilen. Wegen der Ähnlichkeit mit den Döhle-Körpern werden diese auch als "Döhle body like inclusions" bezeichnet, ultrastrukturell handelt es sich aber um parallel liegende Mikrofilamente.
Die molekulargenetischen Veränderungen, die dem Syndrom zugrunde liegen, konnten in jüngster Zeit geklärt werden. Danach muss man von einer Gruppe von MYH9-assoziierten Erkrankungen ("MYH9-related diseases"; Mutation des MYH9-Gens, Chromosom 22q11.2) sprechen. In diese Gruppe gehören die May-Hegglin-Anomalie, das Sebastian-Platelet-Syndrom, das Fechtner-Syndrom und das Epstein-Syndrom.
Die May-Hegglin-Anomalie ist eine familiäre, autosomal dominant vererbte Makrothrombozytopenie mit basophilen spindelförmigen Einschlusskörpern in den Leukozyten. Die Einschlüsse sind gut erkennbar in den Neutrophilen, sie kommen auch in den Eosinophilen und den Monozyten vor, sind in diesen aber kaum erkennbar; auch leichte Leukopenie soll vorkommen können. Eine ausgeprägte hämorrhagische Diathese besteht nicht, die Blutungszeit kann je nach der Schwere der Thrombozytopenie normal oder verlängert sein.
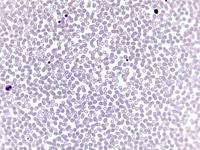
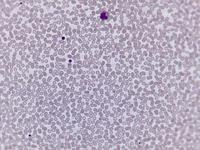
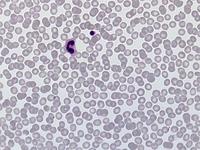
Beim Sebastian-Platelet-Syndrom sind die Einschlusskörper kleiner und sollen nur sichtbar sein, wenn die Blutausstriche innerhalb von vier Stunden nach Blutentnahme gefärbt werden. Eine ausgeprägte hämorrhagische Diathese besteht nicht. Die Thrombozytenzahlen schwanken zwischen 20.000/µl und 100.000/µl.
Beim Fechtner-Syndrom handelt es sich um eine Kombination der Zeichen des Alport-Syndroms (interstitielle Nephritis, Taubheit im Hochtonbereich, Katarakt) mit hämatologischen Veränderungen (Makrothrombozytopenie, Einschlusskörper in den Granulozyten).
Bei dem 1972 beschriebenen Epstein-Syndrom handelt es sich um eine Kombination von hämorrhagischer Diathese bei Makrothrombozytopenie, Innenohrtaubheit und inkonstanter Nephritis.
Bei dem hier dargestellten Fall wurde eine Thrombozytopenie (Werte im Verlauf schwankend zwischen 20.000 und maximal 46.000/µl) abgeklärt. Bei entsprechender Anamnese wurde zunächst auch an eine akute alkoholinduzierte Thrombozytopenie gedacht, die dafür typische rasche Erholung der Thrombozyten unter Alkoholkarenz trat aber nicht ein. Keine Blutungszeichen, anamnestisch keine Blutungsneigung. Bei im Knochenmark gering vermehrten und linksverschobenen Megakaryozyten, Nachweis von vergrößerten Thrombozyten im Blut sowie dem Befund einer nuklearmedizinisch verkürzten Thrombozytenüberlebenszeit (4,7 Tage) mit überwiegendem Abbau in der Milz wurde auch an einen Morbus Werlhof gedacht. Thrombozyten-Antikörper waren negativ.
Die wiederholte Durchmusterung der Granulozyten in Blutbildern ergab keinen zuverlässigen Befund. Zwar ließen sich äußerst selten eben erkennbare basophile Einschlusskörper finden, dieser lichtmikroskopische Befund war jedoch nicht ausreichend für die Diagnose eines hereditären Thrombozytopenie-Syndromes. Die Untersuchung weiterer Angehöriger belegte dann die familiäre Genese. Der eineiige Zwillingsbruder und die Mutter wiesen ebenfalls eine Thrombozytopenie auf. Bei einer zu einem späteren Zeitpunkt durchgeführten elektronenoptischen Untersuchung konnten in den Leukozyten Einschlüsse i.S. eines Sebastian-Platelet-Syndromes nachgewiesen werden.
Literaturreferenzen:
-
Greinacher A, et al. Sebastian platelet syndrome: a new variant of hereditary macrothrombocytopenia with leukocyte inclusions. Blut 1990;61:282-8
[Medline]
-
Mouriquand C, et al. L’anomalie de May-Hegglin. In: Monographies d’hématologie clinique et biologique II. Acquisitions récentes en hématologie. SIMEP editions, Villeurbanne 1971
-
D’Apolito M, et al. Cloning of the murine non-muscle myosin heavy chain IIA gene ortholog of human MYH9 responsible for May-Hegglin, Sebastian, Fechtner, and Epstein syndromes. Gene 2002;286:215-22
[Medline]
-
Kunishima S, et al. Mutations in the NMMHC-A gene cause autosomal dominant macrothrombocytopenia with leukocyte inclusions (May-Hegglin anomaly/Sebastian syndrome). Blood 2001;97:1147-9
[Medline]
-
Yamazaki E, et al. Twenty-one cases of Sebastian platelet syndrom. Rinsho Ketsueki 2001;42:1139-41
[Medline]
-
Epstein CJ, et al. Hereditary macrothrombocytopenia, nephritis and deafness. Am J Med 1972;52:299-310
[Medline]
-
Loreth RM, et al. Sebastian-platelet-Syndrom. Inn Med 1991;18:60-1
-
Begemann H, Rastetter J. (Hrsg.): Klinische Hämatologie. 4. Auflage. Georg Thieme Verlag Stuttgart-New York, 1993
[DNB]
-
Kiefel V, Greinacher A. Differenzialdiagnose und Differenzialtherapie der Thrombozytopenie. Internist 2010;51:1397-410
[Medline]
|

 Zum Seitenanfang Zum Seitenanfang
 Sitemap
| Sitemap
|  |
| 
|
Schwere Thrombozytopenie. Der Hämatologische Notfall. Sysmex Kalender 2008 [ Mehr] Der Hämatologische Notfall. Sysmex Kalender 2008 [ Mehr] |
|
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")
")

